Gold Alloy Nanoparticles in Water
Nong Artrith's Research
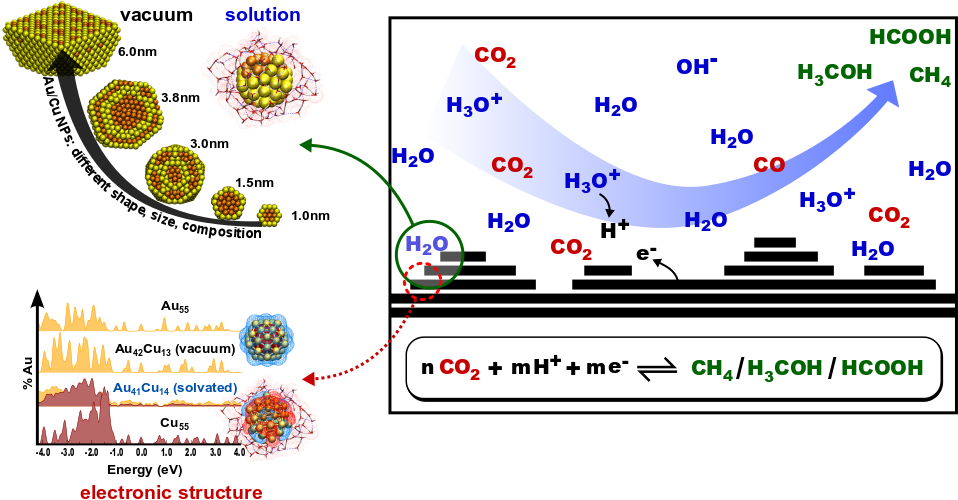
Project Description
Heterogeneous catalytic chemical reactions are at the core of many energy and environment related challenges. Our objective is to understand the catalytic activity at the atomic scale. To achieve this, an accurate and efficient method for predicting the thermodynamically stable compositions, shapes and sizes of catalyst particles under varying conditions is needed. Then the fundamental properties can be determined that make the system active, by examining catalytic behavior on the realistic (non-idealized) catalyst surfaces identified in this way.
We employ a combined approach of atomistic reactive potentials and first principles density-functional theory (DFT). Atomistic potentials allow the simulation of large realistic structures containing thousands of atoms and enable the extensive sampling of the configuration space. Subsequently, the thermodynamic phase diagram of the catalyst structure and stoichiometry can be determined as a function of temperature and chemical potential. Once important structures and possible reactive sites have been identified, DFT calculations of periodic model structures with a smaller number of atoms in the supercell provide the necessary insight into the electronic structure, thereby providing a set of guidelines for predicting and designing novel earth-abundant replacements with comparable or enhanced activity.